La digitalización y la virtualización son cada vez más importantes en una amplia gama de disciplinas científicas. Una de ellas es la ciencia de los materiales: la investigación, el desarrollo y la producción de nuevos materiales dependen en gran medida de la disponibilidad de métodos de simulación rápidos y, al mismo tiempo, precisos.
Esto, a su vez, es beneficioso para un amplio abanico de aplicaciones diferentes: desde los sistemas de almacenamiento de energía eficientes, como los indispensables para el uso de energías renovables, hasta los nuevos medicamentos, para cuyo desarrollo se requiere la comprensión de complejos procesos biológicos. La IA y los métodos de aprendizaje automático pueden llevar las simulaciones en las ciencias de los materiales a un nivel superior.
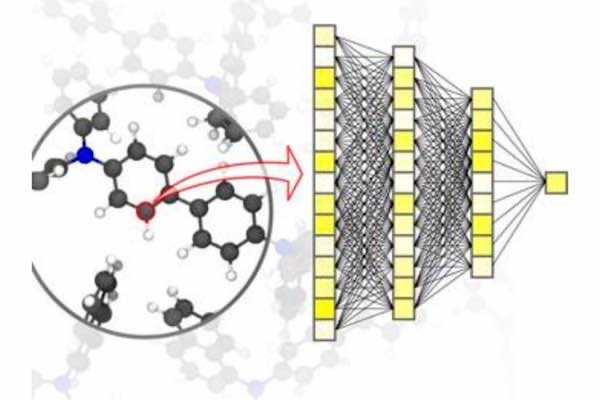
En este artículo, publicado en Nature Materials, se presenta una visión general de los principios básicos del aprendizaje automático utilizado para las simulaciones en las ciencias de los materiales. Esto incluye también el proceso de adquisición de datos y los métodos de aprendizaje.
Los algoritmos de aprendizaje automático no sólo permiten a la inteligencia artificial procesar los datos de entrada, sino también encontrar patrones y correlaciones en grandes conjuntos de datos, aprender de ellos y hacer predicciones y decisiones autónomas. Para las simulaciones en la ciencia de los materiales, es importante lograr una gran precisión en diferentes escalas de tiempo y tamaño, desde el átomo hasta el material, al tiempo que se limitan los costes computacionales.
Por otra parte y para ampliar aún más las posibilidades de las simulaciones de materiales en el futuro, los investigadores sugieren el desarrollo de métodos híbridos: éstos combinan métodos de aprendizaje automático (ML) y de mecánica molecular (MM) dentro del mundo de la química cuántica. Las simulaciones de MM utilizan los llamados campos de fuerza para calcular las fuerzas que actúan sobre cada partícula individual y predecir así los movimientos. Como los potenciales de los métodos ML y MM son bastante similares, es posible una estrecha integración con zonas de transición variables. Estos métodos híbridos podrían acelerar significativamente la simulación de grandes biomoléculas o reacciones enzimáticas en el futuro, por ejemplo.
 710
710
Si el ML os parece un tanto anticuado y a vosotros os va más la Computación Cuántica aquí tenéis una lista de problemas actuales que potencialmente podrían ser resueltos mediante esta última tecnología.
Principalmente desarrolla las posibilidades prácticas que supondría poder simular el funcionamiento de moléculas para buscar mejores baterías, superconductores, fertilizantes o paneles solares.